
Проблемы Эволюции |
Сенсационная книга австралийских иммунологов. Популярно изложено строение иммунной системы позвоночных и механизм образования новых специфических антител методом соматического "гипермутирования" иммуноглобулиновых генов и отбора лимфоцитов. Обоснована гипотеза о возможном механизме наследования приобретенных признаков в иммунной системе. (М.: Мир, 2002. 237 с.)
В гл. 4 мы высказали предположение, что соматические мутации V-генов играли важную роль в эволюции иммунной системы позвоночных. Сформулируем теперь несколько вопросов и постараемся ответить на них. Вопрос первый: каким образом создается картина не-случайности мутаций в ДНК-последовательности, при том, что мутации возникают в результате случайных ошибок? Ответ: по-видимому, существует отбор мутантных ДНК-последовательностей, или их белковых (или РНК) продуктов, сохраняющий только те мутантные гены, которые удовлетворяют селекционному критерию, остальные отбрасываются.
Вопрос второй: каким может быть механизм, предотвращающий появление лишних мутаций, т. е. тех, которые превращают последовательность, уже отобранную как успешную, в плохую? Ответ: по-видимому, существует такой обратный сигнал, который гарантирует, что ген с успешной мутацией не будет мутировать дальше.
И последний вопрос: как обеспечивается мутирование только одного гена? (Большинство генов должно сохраняться неизменным для выполнения надлежащих функций.) Ответ: созданием уникального молекулярного стыковочного устройства, обеспечивающего связывание мутационной машины исключительно с ло-кусом гена-мишени, в котором должны возникать мутации.
С позиций современного неодарвинизма все эти вопросы кажутся вызывающими. Генетики утверждают, что почти все новые мутации вредны. Сейчас эта традиционная точка зрения нуждается в уточнении, так как стал известен особый класс мутаций, которые дают селективные преимущества. Эти мутации возникают в V(D)J-reHax, экспрессирующихся в зрелых В-лимфоцитах. Они обнаруживаются в антителах высокой аффинности, которые образуются примерно через неделю иммунного ответа (табл. 5.1). Такие антитела свойственны долгоживущим В-лимфоцитам памяти. Значит, в результате соматических мутаций появляются антитела, способные более эффективно защищать организм от инфекционного агента. На самом деле описанные выше механизмы «работали» в течение миллионов лет и были результатом биологического процесса «проб и ошибок» во взаимодействии молекул.
Таблица 5.1.
Основные понятия, относящиеся к процессам соматического гипермутирования и обратной связи генов сомы и зародышевой линии
• RT-мутаторсома
(RT-mutatorsome)Гипотетическая молекулярная органелла в ядре В-клетки, ответственная за соматическое гипермугирование (RT = обратная транскриптаза, reverse transcriptase). См. рис. 5.6
• Апоптоз
Биологически запрограммированная гибель клеток
• Гибридома
Продукт слияния нормальной В-клетки с непрерывно делящейся опухолевой В-клет-кой. Такая клетка ведет себя как раковая и может независимо расти в культуре ткани. Она секретирует антитела только одной специфичности (исходной В-клетки)
• Гистоны
В клетках эукариот очень длинные нити ДНК имеют вид упорядоченных петель, связа-ных с белками гистонами, что обеспечивает упаковку ДНК в небольшом пространстве ядра
• Конфигурация зародышевой линии
Структура ДНК-последовательности вариабельных, или V-элементов генов зародышевой линии. которые не подверглись перестройке с J или D/J элементами. Генетический символ V(D)J
• Локус-специфическое устройство
Термин используется для обозначения участка стыковки (связывания) RТ-мутаторсомы. Он позволяет ограничить соматические гипермутации только V(D)J и ближайшими ДНК-последовательностями
• Матричная молекула
Любая одноцепочечная ДНК- или РНК-последовательность, которая служит матрицей для создания копии с помощью репликазы
• мРНК
Информационная РНК, которая транскрибируется (копируется) с ДНК-последовательности гена
• Мутагены
Химические вещества или проникающая электромагнитная радиация, способные вызывать мутации
• Обратная связь сомы и зародышевой линии
Процесс, в ходе которого соматически мутированные V(D)J-reны обратно транскрибируются в кДНК, и эти копии рекомбинируют с гомологичной последовательностью V-гена половой клетки так, что замещают ее
• Обратная транскриптаза
Фермент, который копирует РНК-последовательность в ДНК-последовательность
• Перестроенный ген варибельной области
Сокращенное обозначение V(D)J, применимое к перестроенным генам вариабельных областей Н-цепи (VDJ) и L-цепи (VJ)
• Репликаза/полимераза
Общее название ферментов, которые создают дочерние копии ДНК- или РНК-последовательностей. К ним относятся ДНК-полимераза (осуществляет репликацию ДНК), РНК-полимераза (осуществляет транскрипцию) и обратная транскриптаза (создает ДНК-последовательность по матрице РНК)
• РНК-посредник
Этот термин отражает тот факт, что копия гена (ДНК-последовательность), превращенная (в процессе транскрипции) в РНК, может обратно транскрибироваться в кДНК, или ретротранскрипт
• Созревание аффинности
Мутантные антитела, продуцируемые В-клетками памяти, которые возникают в центрах размножения, имеют более высокую аффинность (сродство) к антигену, чем антитела на ранних стадиях иммунного ответа
• Соматическая конфигурация
Перестроенный V-ген вариабельной области,
который обнаруживается только в зрелых В- и Т-лимфоцитах. Генетический символ V(D)J
• Соматическое гипермугирование
Мутационный процесс, вызванный стимуляцией В-клетки антигеном, затрагивающий перестроенный ген вариабельной области антитела V(D)J зрелой В-клетки в центре размножения
• Точковая мутация
Замена одного основания. Например, замена основания G на любое из трех других оснований Т, А и С (рис. 2.5)
• Центр размножения
Особая область в лимфатических узлах или селезенке, в которой обнаруживается интенсивная пролиферация (деление) и гибель (апоптоз) клеток. Большинство клеток в таком центре - это В-лимфоциты. Центры размножения являются местами соматического гипермутирования и созревания аффинности, вызываемых антигеном
25 лет назад словосочетание «соматическая мутация» вызывало горячий спор среди иммунологов. В последующие годы споры стали менее оживленными, а многочисленные молекулярные исследования, показали реальность высокого уровня вызываемых антигеном соматических мутаций. Теперь этот процесс называется «соматическим гипермутированием» генов вариабельной области антитела, V(D)J.
Интересно, что в недавних экспериментах, выполненных под руководством Нобелевского лауреата Ролфа Цинкернагеля, было показано, что при иммунном ответе на некоторые вирусные белки не обнаруживается созревания аффинности, основанного на соматическом гипермутировании. Кроме того, оказалось, что антитела высокой аффинности не лучше освобождают организм от вирусной инфекции, чем антитела низкой аффинности. Между тем сейчас считается, что соматическое гипермутирование необходимо для создания антител высокой аффинности, которые лучше защищают от инфекции. Если результаты группы Цинкернагеля подтвердятся для более широкого спектра инфекций и для других позвоночных, это будет означать, что оптимально функционирующие антитела могут быть отобраны из предсуществующего репертуара, и нет необходимости в мутациях. То есть, для ныне живущих позвоночных соматическое гипермутирование и созревание аффинности, возможно, излишни.
Однако в начале эволюционного развития системы приобретенного иммунитета, когда репертуар V-генов клеток зародышевой линии был много меньше, чем у срвременньис позвоночных (он должен был стартовать с одного гена), соматическое гипермутирование могло давать огромное селективное преимущество. Этот процесс мог обеспечивать формирование чрезвычайно широкого репертуара антител, экспрессирующихся в В-лимфоцитах в течение жизни одного животного. Далее, при существовании обратной связи сомы и зародышевой линии соматические мутации генов вариабельной области антител могли бы стать решающим инструментом эволюции, ускоряющим построение репертуара V-генов половых клеток. Все доступные данные согласуются с этим предположением. Соматическое гипермутирование обнаруживается у всех челюстных позвоночных, имеющих систему приобретенного иммунитета, включая наиболее примитивных — хрящевых рыб.
Явление соматического гипермутирования наиболее ярко продемонстрировано для В-лимфоцитов мыши, аналогичный процесс имеет место и у человека. У других позвоночных (кур, кроликов и овец) также наблюдается гипермутирование в В-лимфоцитах, хотя и несколько иное. Эксперименты по изучению механизма соматического гипермутирования проводились в основном на генетически модифицированных (трансгенных) мышах, в частности, в лаборатории Кембриджского университета под руководством Нобелевского лауреата Цезара Мильштейна (Milstein) и его сотрудника Майкла Нойбергера (Neuberger). Эти методы еще не применялись к другим видам.
В этой главе мы сначала рассмотрим, как возникают генные мутации вообще, а затем остановимся на процессе соматического гипермутирования генов V(D)J. Мы покажем, что это строго регулируемый процесс, в нем участвует «локус-специ-фичное устройство», которое ограничивает мутирование ДНК-последовательностью перестроенного V(D)J-yчастка, защищая гены константной области (С) и все другие гены в гено-ме от вредного влияния случайных мутаций.
Мутации возникают в склонных к ошибкам процессах копирования, включающих РНК-посредника
Мутацией называется изменение последовательности нуклеотидов в ДНК (рис. 2.6). Если мутация происходит в той области ДНК, которая кодирует белок, она изменяет триплетный кодон и может привести к замене аминокислоты, определяемой этим код оном. Таким образом, появление другой аминокислоты в белковой цепочке может быть вызвано изменением одного единственного основания в ДНК-последовательности (точковая мутация). Большая часть измененных белков функционирует ненормально (хотя иногда они и выполняют совершенно иную функцию, на ином физиологическом или метаболическом фоне). Поэтому с точки зрения дарвиновского «выживания наиболее приспособленного» большинство мутаций вредны и ставят клетку или многоклеточный организм в неблагоприятные условия при естественном отборе.

Рис. 5.1. Схема, показывающая как точковая мутация может привести к образованию нового мутантного белка с иной третичной структурой. Примечание: Дополнительную информацию см. в табл. 1.1, 5.1 и на рис. 2.5, 2.6 и 2.7.
Рис. 5.1 иллюстрирует эти рассуждения. На нем показаны нормальный и измененный белки. Обратите внимание, что одна единственная точковая мутация G -> С в кодоне, определяющем аспарагиновую кислоту (Asp), дает начало новому кодону, определяющему гистидин (His), в том же положении белковой цепочки (см. также приложение). Такая мутация может привести к радикальным последствиям для мутантного белка, который, складываясь, приобретает другую форму и, следовательно, — другую функцию (т. е. нормальная функция может быть утрачена). Этот принцип был продемонстрирован 40 лет назад, когда определили последовательность аминокислот бета-цепи гемоглобина здоровых людей и людей, страдающих серповидно-клеточной анемией. При этом наследственном заболевании нарушена способность гемоглобина переносить кислород. Молекула гемоглобина — гетеродимер, состоящий из двух альфа-цепей, или альфа-субъединиц, и двух бета-цепей, или бета-субъединиц. Заболевание проявляется у гомозиготных индивидов, у которых обе гомологичные хромосомы несут дефектный ген бета-цепи. Гетерозиготы, которые имеют одну нормальную и одну мутантную копию гена бета-цепи, болеют в легкой форме (так как половина их молекул гемоглобина способна нормально переносить кислород). Оказалось, что у больных серповидноклеточной анемией в 6-м положении бета-цепи вместо глутаминовой кислоты находится валин. И эта единственная замена в цепи из 146 аминокислот приводит к образованию больших агрегатов мутантных молекул гемоглобина в эритроцитах, деформирующих клетку так, что она принимает форму серпа. Интересно, что ген серповидноклеточной анемии сохраняется в Африке, потому что гетеро-зиготы имеют селективное преимущество при заражении малярией, так как плазмодий не может размножаться в серповидных эритроцитах столь же эффективно, как в нормальных.
Это опять возвращает нас к ключевому вопросу: как возникают генные мутации? Еще не так давно считалось, что они возникают «спонтанно» под влиянием космических лучей, рентгеновского излучения или ультрафиолетового света. Чарлз Дарвин называл такого рода изменения «спортами» и предполагал, что их случайно порождают условия Природы. Случай действительно играет роль, — разные мутации появляются с разной частотой. Однако сейчас, после 30—40-летнего периода накопления данных по вирусологии и молекулярной биологии, тайн вокруг причин появления мутаций гораздо меньше. В начале 1980-х гг. Дарил Ренни (Renney) из Университета Ла Троуб (Мельбурн) провел очень полезный анализ этой проблемы [7]. Благодаря его работе и выполненным ранее исследованиям Нобелевских лауреатов Артура Корнберга (Komberg), Манфреда Айгена и Говарда Темина (открывшего обратную транскрипцию у РНК-содержащих опухолевых вирусов) и вирусолога Джона Холленда (Holland), мы имеем логически последовательный способ анализа механизмов возникновения генных мутаций. Все дело в точности копирования ДНК- или РНК-последовательностей по матричным молекулам ДНК или РНК, которые осуществляются четырьмя ферментами, копирующими нуклеиновые кислоты: ДНК-полимеразой, РНК-полимера-зой, РНК-репликазой и обратной транскриптазой.
Исследования на молекулярном уровне показали, что ферменты, участвующие в репликации ДНК, способны к редактированию и исправлению ошибок. Возникновение мутаций в ходе репликации ДНК — редкое событие (рис. 5.2). Максимальная частота таких мутаций, вероятно, меньше, чем 10-8, а истинная частота ошибок, вероятно, еще меньше — около 10-10 (меньше, чем одна на 10 миллиардов реплицированных оснований). Чрезвычайно высокая точность копирования информации обеспечивается ДНК-полимеразой («машиной, копирующей ДНК»), которая по мере продвижения вдоль матричной ДНК-цепи проверяет, нет ли ошибок во вновь синтезированной копии. О наличии ошибок она «узнает» по искажению двойной спирали ДНК, которое имеет место, если Т соединится с G или С с А. Обнаружив такой участок, ДНК-полимеразный ферментный комплекс вырезает неправильное основание (или группу оснований) и вставляет то, которое должно быть на этом месте (законное основание). Скорость точной репликации у бактерий примерно 500 оснований в секунду, а у высших клеток (включая клетки человека) около 50 оснований в секунду. ДНК хромосом высших клеток много длиннее, а сами хромосомы устроены намного сложнее, чем маленькие и простые бактериальные геномы. У высших клеток, в отличие от бактерий, ДНК в хромосомах образует комплекс с белками (гистонами), которые участвуют в сворачивании длинных нитей ДНК в серию петель, для того чтобы их можно было упаковать внутри ядра. Репликация ДНК начинается одновременно в нескольких сайтах (точках) каждой хромосомы, поэтому большой набор ДНК-последовательностей реплицируется за 5—20 ч.

Рис. 5.2. Частота ошибок при синтезе ДНК и РНК. Примечание: о частоте ошибок судят по частоте включения неправильного основания на одно основание за одно событие копирования (см. также рис. 2.4); дц = двухцепочечная, оц = одноцепочечная.
Вспомним, что в гл. 2 мы уже обсуждали высокий уровень ошибок при образовании РНК по матрице ДНК (транскрипции) и при образовании ДНК по матрице РНК (обратной транскрипции). Оба этих типа копирования характеризуются частотой точковых мутаций Ю-3—Ю-4, что существенно выше, чем частота ошибок при репликации ДНК (от Ю-8 до Ю-9). Неточность, большое число ошибок имеют место и при репликации генома РНК-содержащих вирусов, например, вируса гриппа. Этим объясняется быстрое генетическое изменение вируса, приводящее к пандемиям гриппа. В жизненном цикле вируса СПИДа (ВИЧ) чередуются неточные процессы копирования РНК -> ДНК (на стадии интеграции) и ДНК -> РНК (на стадии экспрессии в течение инфекционного цикла). Для этого вируса также характерна высокая частота мутаций. Таким образом, все процессы копирования, включающие одноцепочечные РНК-посредники (превращение РНК в ДНК и наоборот), идут с большим числом ошибок, при этом репарация последовательности невозможна, поскольку ферменты, осуществляющие такое неточное копирование полинуклеотидов (РНК-полимераза, обратная транс -криптаза и РНК-репликаза), как оказалось, не имеют функций проверки и исправления ошибок.
Все сказанное выше означает, что какая-то доля мутаций в ДНК может возникать в результате ошибок копирования, включающего промежуточные РНК-посредники (рис. 5.2).
Мутации, которые передаются потомкам, появляются с низкой частотой. Они возникают в половых клетках и называются генеративными. Это редкие ошибки, которым удалось ускользнуть от «проверок» ДНК-полимеразы во время репликации ДНК и упаковки ее в гаметы самцов и самок (сперматозоиды и яйцеклетки). Они могут вызывать дефекты (т. е. влиять на фенотип и состояние здоровья индивида). Например, в случае серповидцоклеточной анемии такая мутация обуславливает тяжелую патологию у гомозигот (у которых обе копии гена дефектны) и более легкую форму болезни у гетерозигот, потому что белковый продукт нормальной копии гена частично компенсирует отрицательный эффект дефектной копии.
Однако существуют варианты некоторых генов (альтернативные формы генов называют аллелями), которые не влияют на здоровье индивида, у которого они проявляются. Эти варианты составляют нормальную изменчивость в популяциях организмов, существующую, по предположению Дарвина, до того, как начинает действовать естественный отбор. Важный вопрос: как появляются эти «добрые» аллели?. Согласно неодарвинистским представлениям, все эти аллели возникли в результате случайных мутаций в ДНК зародышевой линии и сохранились в популяции (так называемом «пуле генов») вследствие естественного отбора. В гл. 7 мы постараемся дать альтернативное объяснение этого феномена в рамках теории обратной связи соматических и половых клеток.
Предполагают, что рост числа врожденных аномалий и спонтанных абортов вызван факторами окружающей среды, такими как загрязнение токсическими химическими веществами. Например, резкое повышение частоты врожденных аномалий зарегистрировано в городах, расположенных вокруг сильно загрязненного, гибнущего Аральского моря. Такие же данные имеются относительно ветеранов вьетнамской войны и жителей северного Вьетнама, подвергшихся воздействию токсичных дефолиантов. Вещества, которые действуют на гены, изменяя кодирующую ДНК-последовательность, называются мутаге-нами. Возможно, их действие основано на том, что они нарушают нормальный процесс репарации. Установлено, что в клетках бактерий и эукариот, в которых индуцировано большое число повреждений ДНК, включается склонная к ошибкам репарация.
Многоклеточный организм состоит из сотен миллионов клеток, некоторые из них непрерывно и быстро делятся. Например, у человека и других позвоночных все клетки крови обновляются со скоростью десять миллионов в день. Эпителиальные клетки кожи и слизистых (пищеварительного тракта и носоглотки) ежедневно образуют миллионы дочерних клеток, замещающих израсходованные, т. е. те клетки, которые слущиваются с эпителиальных поверхностей. Во внутренних органах (сердце, печени, почках и мозге) скорость замещения клеток низкая. Нейроны (нервные клетки) у взрослого человека не делятся вообще. При делении клетки ДНК в ядре удваивается, образуются копии всех хромосом, которые передаются дочерним клеткам. Если это происходит в большом числе клеток, надо ожидать появления какого-то числа соматических мутаций (несмотря даже на то, что частота мутаций при репликации ДНК низка). Следовательно, у крупных многоклеточных животных соматические мутации будут появляться все время, особенно в тех клеточных популяциях или тканях, где скорость замещения клеток очень высока.
Все виды злокачественных опухолей вызваны соматическими генными мутациями, которые делают клетки невосприимчивыми к сигналам, ограничивающим их рост или вызывающим гибель; такие клетки начинают «жить своей собственной жизнью». Они могут мутировать дальше, становясь локально агрессивными или давая начало метастазам. Наглядный пример последствия соматических мутаций — кожные формы рака. Они возникают из единичной мутантной клетки при ее делении. Клон клеток, подобно колонии плесневых грибов на черством хлебе, растет на ограниченном участке кожи. Наиболее злокачественная форма рака кожи — меланома (пигментированная опухоль). Сейчас известно, что ее образование провоцирует ультрафиолетовое излучение солнца.
Соматические мутации, приводящие к раку, как и мутации, неблагоприятно действующие на функции важных структурных белков и ферментов, несомненно вредны. Мы кратко рассказали о них, чтобы показать контраст с полезными мутациями, которые происходят в генах вариабельных областей антитела, и подчеркнуть исключительность процесса контроля/отбора в мутантных В-лимфоцитах. Теперь повторим наши ключевые вопросы и попытаемся установить возможные биологические механизмы, обеспечивающие эти процессы.
Сейчас известно, что в ходе иммунного ответа в перестроенных V(D)J-генax соматические мутации возникают с высокой частотой. В отобранных антигеном В-клетках частота мутаций V(D)J-генов составляет примерно 1/1000 — 1/10000 оснований на репликационное событие. Это в миллионы раз выше, чем частота мутирования генов, передающихся с половыми клетками. Мутантные V-области антитела появляются через 5—10 дней после воздействия антигена. Считается, что увеличение аффинности (сродства к антигену) антитела основано на соматическом мутировании и отборе в течение иммунного ответа (рис. 3.8). Несмотря на то, что молекулярные механизмы мутационного процесса в соматических клетках иммунной системы активно исследуются во многих лабораториях мира, включая и лабораторию Теда Стила и Боба Бландэна, еще остаются вопросы, требующие объяснения. Может ли биологическая система обеспечивать полезность соматических мутаций (например, удаляя вредные мутации, но сохраняя полезные)? По нашему мнению, ответ на этот вопрос — да, может. Иммунная система выработала два тесно связанных процесса — соматического мутирования и отбора наиболее приспособленных, которые обеспечивают животным потенциальные преимущества.
Прежде, чем перейти к молекулярным и клеточным деталям процесса соматического мутирования, надо описать тонкую структуру вариабельной области, которая образует антигенсвязывающий центр антитела (ТкР имеют сходную структуру).
Почти 30 лет назад Элвин Кэбот (Kabat) с коллегами начали определять аминокислотные последовательности вариа-бельных областей антител человека. По мере того как накапливалась информация, Т. By (Wu) и Э. Кэбот смогли сравнить последовательности вариабельных областей тяжелых и легких цепей и построить график изменчивости аминокислот в каждом положении белковой цепи. Их результат, который называется графиком Ву—Кэбота, приведен на рис. 5.3. На этом графике выявляется три гипервариабельных участка, другие участки называются каркасными (FR, от англ. framework region). Гипервариабельные участки образуют поверхность, которая вступает в тесный контакт с поверхностью антигена. Каркасные участки составляют структурный скелет, или остов, V-областей антигенсвязывающего центра. В зависимости от природы антигена, поверхность антитела может быть разной: от относительно плоской с небольшими углублениями или выступами до глубоких «карманов», которые вмещают выступы поверхности антигена. Поверхность антитела высокой аффинности комплементарна поверхности антигена.
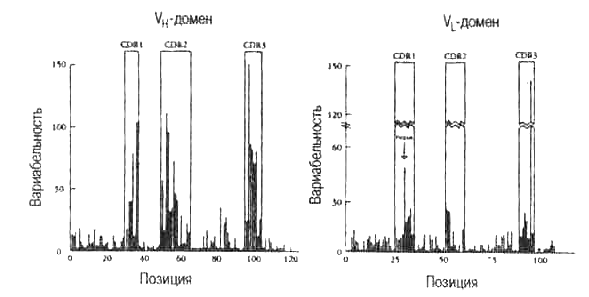
Рис. 5.3. График Ву-Кэбота.
График Ву—Кэбота показывает, что гипервариабельные участки вариабельной области Н- и L-цепей антител совпадают с антигенсвязываю-щими центрами, названными участками, определяющими комплементарность
В большом наборе антител человека разной специфичности относительную вариабельность рассчитывают для каждой из 100 позиций аминокислот вариабельной области тяжелых и легких цепей. Этот график показывает, что вариабельность не одинакова — выявляются три участка гипервариабельности (HV), между ними расположены участки с меньшей вариабельностью, названные каркасными сегментами (FR). Известно, что HV-участки совпадают с теми районами белковой цепи, которые вступают в непосредственный контакт с эпитопами антигена. Эти участки называются участками, определяющими комплементарность, или CDR. Нумерация начинается с левого конца молекулы (рис. 3.2), так что CDR3 расположен рядом с константной областью. Высоко неслучайная картина вариабельности, продемонстрированная By и Кэботом, свидетельствует о том, что вариабельные последовательности подвергаются положительному дарвиновскому отбору связыванием антигеном на уровне HL-гетеродимеров. Семейство V-генов всех позвоночных дает картину вариабельности Ву—Кэбота. См. табл. 3.1 и 5.1. (По J. Kuby. Immunology, 3rd edition. W. Н. Freeman & Co., 1997. Перепечатано с разрешения издателя. Основано на Е. A. Kabat et al. 1977. Sequence of Immunoglobulin Chains, US Department of Health Education and Welfare.)
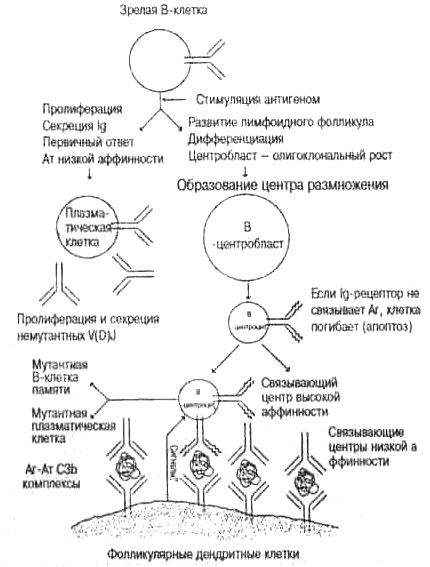
Рис. 5.4. Соматическое мутирование и отбор мутантных клеток в центре размножения.
После отбора антигеном циркулирующей В-клетки активированная клетка будет или активнее секретировать антитело, или станет клеткой-основателем в центре размножения, где происходит мутирование V(D)J-reHOB вариабельной области. Антитела, образованные в начале ответа, формируют комплексы антиген — антитело, которые связываются с поверхностью фолликулярных дендритных клеток (ФДК). Эти анти-генпрезентирующие клетки образуют в центре размножения широкую мембранную сеть. После фазы быстрой пролиферации клеток (В-центробластов) меньшие по размеру неделящиеся В-центроциты проверяют свое мутантное антитело на связывание с антигеном, проявляющимся на ФДК. Если мутантное антитело не функционально или имеет низкую аффинность, клетка погибнет вследствие программированной гибели (апоптоза). Если у мутанта более высокая аффинность, чем у антитела, связанного с антигеном на поверхности ФДК, он вытесняет это антитело и связывает антиген. Это дает сигнал В-клетке, защищающий ее от гибели. Отобранная мутантная В-клетка покидает центр размножения и становится или плазматической клеткой (образующей и секретирующей антитела высокой аффинности), или долгоживущей клеткой памяти. Соматические мутанты данной У(0)и-последовательности аминокислот, проанализированные как на рис. 5.3, дают структуру By—Кэбота, свидетельствующую о том, что вариабельные области антител отбираются связыванием антигеном. (Перепечатано из Steele E. J. et al. Immuno-logical Reviews, vol. 135: 5-49,1993 с разрешения издателя Munksgaard International Publishers, Copenhagen.) Примечание: Ат — антитело, Аг — антиген, СЗb — активированный белок крови, который связывает Ат-Аг комплекс.
График Ву—Кэбота, следовательно, можно считать распределением аминокислот в вариабельных областях «успешных» антител, которые хорошо связываются с антигеном. В более поздних работах был построен график By—Кэбота для антител, которые возникли в результате соматических мутаций одной V-последовательности зародышевой линии в ответ на один антиген. Этот график ясно показывает, что изменчивость, появляющаяся в вариабельной области антитела, не случайна.
Если для какого-то набора данных о последовательностях аминокислот вариабельной V(D)J области антитела получен график, подобный приведенному на рис. 5.3 (мы будем называть его структурой By—Кэбота), можно считать, что есть серьезные указания на антигенсвязывающий отбор HL-гетеродиме-ров антитела. Другими словами, структура By—Кэбота (наличие на графике пиков изменчивости в районах контакта с антигеном) является показателем того, что изменчивость аминокислотных последовательностей в изучаемой популяции V(D)J-no-следовательностей создана антигенсвязывающим отбором. Мы вернемся к этому важному заключению в следующей главе, когда попытаемся объяснить, почему структура By—Кэбота наблюдается в популяциях неперестроенных V-элементов половых клеток.
Напомним (рис. 4.5), что полный ген вариабельной области является соматической конструкцией из V-, D- и J- элементов в тяжелой Н-цепи и V- и J-элементов в легкой L-цепи, соединенных в результате перестройки ДНК в В-клетках. Два участка гипервариабельности закодированы в V-элементе зародышевой линии, а третий, который затрагивает соединенные V-D-J или V-J перестройки, никогда не обнаруживается в зародышевой линии позвоночных, хотя есть исключения — у хрящевых рыб (скатов, рогатых акул) и V-псевдогенов курицы. Третий гипервариабельный участок очень важен для распознавания антигенов. Антитело, у которого нет этого участка (имеющее только два кодируемых в зародышевой линии гипервариабельных участка), не способно связывать антиген. Следовательно, V-гены зародышевой линии не могут подвергаться прямому дарвиновскому отбору. Отбор опосредован и должен затрагивать V-D-J-и V-J-перестройки, которые происходят только в соме.
Все имеющиеся данные говорят о том, что в В-лимфоцитах мутируют только перестроенные V(D)J-гены, кодирующие белок антитела. Другими словами, вариабельные гены, остающиеся в конфигурации зародышевой линии, т. е. неперестроенными, не накапливают соматических мутаций. Следовательно, первым этапом контроля, позволяющим появляться только «благоприятным мутациям», является перестройка ДНК, в результате которой образуется V(D)J-мишень, на которую действует мутатор. Следующий этап контроля — это сама мутационная машина, которая должна связываться с уникальной структурой, ассоциированной с перестроенной V(D)J-последовательностью и ограничивающей мутирование только этим участком ДНК.
Надо отметить еще один важный момент. Процесс соматического гипермутирования у мышей и человека вызван стимуляцией антигеном. У других видов (например, овцы) соматический мутагенез в специализированных лимфоидных тканях, связанных с кишечником, включается другими сигналами (они пока неизвестны). Таким образом, изменчивость V(D)J-генов лимфоцитов вызвана влиянием окружающей среды или адаптивным сигналом. Это означает, что в ходе развития иммунной системы организма генетическая изменчивость может быть вызвана средой. Это противоречит неодарвинистской догме о том, что вся изменчивость генов зародышевой линии предсуществует до того, как начинает действовать отбор. Решающий эксперимент, доказывающий этот важный момент, был проведен Урсулой Сторб (Storb) с коллегами в середине 1980-х гг. Авторы ввели ограниченное число копий известного перестроенного гена легкой цепи (VJ) в ДНК зародышевой линии инбредных мышей (такие животные называются трансгенными). Им удалось показать, что у неиммунизированных трансгенных мышей VJ-трансген не мутировал: последовательность нуклеотидов этого участка ДНК, выделенного из В-лимфоцитов селезенки трансгенных мышей, не изменилась. Однако при иммунизации антигеном, который связывает антитела, кодируемые этой VJ-последовательностью, VJ-трансген накапливал многочисленные соматические мутации.
Где происходят соматические мутации? Могут ли они возникать в подвижных В-клетках в крови и лимфе, или они происходят в каких-то особых местах? В середине 1980-х гг. Дэвид Грей (Gray) и Йан Макленнан (MacLennan), работающие в Университете Бирмингема, предположили, что соматические мутации происходят в специализированных постантигенных образованиях, называемых центрами размножения. Такие участки интенсивного деления (пролиферации), появляющиеся после стимуляции антигеном в лимфатических узлах и селезенке, давно известны. Также было известно, что 80% В-клеток в центре размножения (или даже больше), по-видимому, там же и погибают. В 1960-х гг. Гас Носсал (Nossal) и Гордон Эйда, работающие в Медицинском центре Уолтера и Элайзы Холл, используя радиоактивно меченные белковые антигены установили, что недеградированные формы антигена, связанные с антителами, располагаются на поверхностной мембране клеток в центре размножения (и могут оставаться там в течение многих месяцев). Комплексы антиген—антитело располагаются на поверхности крупных специализированных клеток, выросты клеточной мембраны (дендритные выросты) которых распространяются по всему центру размножения. Эти клетки называются фолликулярными дендритными клетками (ФДК).
Фолликулярные дендритные клетки в центре размножения взаимодействуют с популяцией В-клеток (рис. 5.4). Грей и Макленнан предположили, что соматические мутации возникают в быстро делящихся В-клетках центра размножения. «Успешные» мутанты, способные связываться с антигеном и конкурировать за его молекулы, которые проявляются в комплексе антиген—антитело на ФДК, выживают, остальные гибнут. В то время это было очень смелое предположение, объясняющее созревание аффинности. В нем объединялись быстрое соматическое мутирование и связанный с антигеном процесс отбора, спасающий от гибели тех мутантов, которые успешно конкурируют за антиген на ФДК в силу высокой аффинности нового мутантного антитела.
За пять лет группа под руководством Гарнета Келсо, Клауса Раевского (Rajewsky) и Клаудии Берек (Berek) доказала правильность некоторых предсказаний Грея-Макленнана. Оказалось, что соматические мутации перестроенных V(D)J-генов действительно происходят в В-клетках центра размножения. Келсо и Раевский пошли еще дальше, продемонстрировав, что в делящихся В-клетках, которые не были в центре размножения, V(D)J-участок не мутирует. Это означает, что критический сигнал, активирующий «мутатор», находится в центре размножения. Примерно в то же время Йан Макленнан и его группа установили, что, если у потомков В-клетки особой обработкой вызвать сшивки мутантных Ig-молекул (этот процесс имитирует связывание антигена), то такие клетки не умирают.
Итак, к 1991 г. центры размножения представлялись как участки лимфоидной ткани, в которых активно идет соматическое гипермутирование — регулируемый процесс, связанный с анти-гензависимым отбором. Мы хотим подчеркнуть, что центр размножения и у человека, и у мыши развивается только в результате развития иммунного ответа на чужеродный антиген. Важнейшие события, происходящие в центрах размножения, приведены на рис 5.4.
Экспериментальные данные позволяют воссоздать картину возникновения центра размножения во времени и пространстве. В кровотоке в результате связывания с чужеродным антигеном происходит отбор В-клетки. После этого она мигрирует в лимфоидные зоны селезенки или лимфатических узлов. Затем, получив особый сигнал от хелперной Т-клетки, она начинает делиться. Эти активированные В-клетки продуцируют антитела, которые образуют комплексы антиген-антитело на поверхности фолликулярных дендритных клеток (ФДК). Несколько потомков исходной отобранной В-клетки заселяют особые участки лимфоидной ткани, содержащие ФДК, которые называются первичными фолликулами. Одна или несколько из этих клеток-основателей начинают быстро делиться (на рис. 5.4 они названы В-центробластами); на их поверхности нет антител.
Фаза пролиферации (деление клеток каждые пять-семь минут в течение примерно пяти дней) приводит к образованию популяции численностью порядка 20 тысяч дочерних клеток, которые называются центроцитами. Они прекращают деления, и на их поверхностной мембране снова появляются антитела. На каком-то этапе образования популяции центроцитов происходит гипермутирование перестроенных V(D)J-генов. Теперь центр размножения созрел, и сложная постантигенная структура содержит В-центроциты, хелперные Т-клетки и ФДК. Последние образуют развитую сеть комплексов антиген—антитело, включающих антитела, синтезированные в первые дни ответа (см. рис. 5.4).
Затем, по-видимому, происходят следующие события. Во-первых, тысячи центроцитов составляют гигантский репертуар клеток, поверхностные антитела которых кодируются соматическими мутациями. Большинство этих антител (примерно 80%) не способны связывать антиген. Как и для любых других белков, большинство мутаций приводит к изменению формы антитела, а это нарушает соответствие форме антигена. Однако некоторые редкие мутации могут приводить к антителам, лучше соответствующим форме антигена, чем исходные (т. е. с более высокой аффинностью). Новые антитела расположены на поверхности В-центроцитов и могут конкурировать за молекулы антигена, расположенные в комплексах антиген—антитело на поверхности фолликулярных дендритных клеток. Однако для того, чтобы успешно конкурировать с антителом из комплекса (образованным в первые дни ответа), новое мутантное антитело должно иметь ту же или большую аффинность. Вот суть механизма созревания аффинности — конкурентный антигенсвязы-вающий отбор. Центр размножения — это недолговечный «орган селекции и разведения» V(D)J-генов, где выживают только наиболее приспособленные В-клетки. Неудачные (с низкой аффинностью, нефункциональные) мутантные В-клетки (а их большинство) исчезают в результате запрограммированной клеточной гибели, которая называется апоптозом.
Итак, клональная селекция В-клеток происходит в два этапа. На первом чужеродный антиген отбирает В-лимфоцит из популяции разнообразных клеток. Затем эти В-центробласты быстро делятся, образуя популяцию в 20 тысяч новых В-клеток, имеющих мутантные вариабельные V(D)J-области. На втором этапе мутантные линии В-клеток с высокой аффинностью отбираются антигеном, выживают, делятся и секретируют антитела высокой аффинности или становятся долгоживущими клетками памяти. В этом нет ничего мистического. Это традиционный дарвиновский отбор, действующий в большой изменчивой клеточной популяции, из которой выживают немногие — только те, которые продуцируют мутантные антитела с наивысшей аффинностью к антигену. Центр размножения, следовательно, — котел, в котором «варится» генетическая гипервариа-бельность. Однако все процессы в нем строго регулируются и контролируются. V(D)J-гены (только эти гены, а не другие) В-лимфоцитов мутируют в высоко специализированной и регулируемой среде центра размножения.
До сих пор мы подчеркивали значение дарвиновского естественного отбора в иммунной системе. Однако мы ввели и определенно неоламаркистскую идею, а именно, роль сигнала внешней среды (антигена) в создании генетического разнообразия, на которое действует естественный отбор. Может ли этот ключевой эволюционный вывод вызвать такие же споры, которые были вызваны идеей «разнообразия, вызванного антигеном», впервые предложенной Элистэром Каннингемом в середине 1970-х гг.?
Примерно 25 лет назад Элистэр Каннингем, работающий в Центре медицинских исследований Джона Куртина в Канберре, впервые высказал идею о соматическом мутировании вари-абельных генов антител, вызванном антигеном. Он также первым провел эксперименты, демонстрирующие это явление. Примерно в то же время исследовательские группы Мелвила Кона (Институт Салк, Сан-Диего, США) и Сусуми Тонегавы (Базельский институт иммунологии, Швейцария) получили молекулярные доказательства существования соматического мутирования (путем анализа последовательностей белка и ДНК). Оба эти открытия были сделаны в разгар споров между сторонниками идеи соматического разнообразия и идеи разнообразия, закодированного в зародышевой линии. Самая первая гипотеза о молекулярном механизме соматического мутирования Сиднея Бреннера и Цезара Мильштейна (Кэмбриджский университет, Англия) подразумевала существование некоего склонного к ошибкам типа репликации ДНК. Предполагалось, что высоко точное копирование матрицы ДНК нарушается при репликации генов вариабельной области антител. (Это нарушение объяснялось неспособностью ДНК-полимера-зы в данном случае исправлять ошибки.) Таким образом, первые модели соматического мутирования основывались на репликации ДНК и делении клетки: так как активированные антигеном В-клетки быстро делятся, появляются мутанты, у которых изменена ДНК, кодирующая вариабельную область.
Подобно всем новым научным гипотезам, экспериментам и интерпретациям пионерская работа Элистэра Каннингема вызвала споры. Однако к 1981 г. поток публикаций из лабораторий Патрисии Гирхарт (Gearhart), Урсулы Сторб, Эла Ботуэлла (Bothwell), Лероя Худа (Hood), Дэвида Балтимора и Клауса Раевского подтвердил правильность основного предположения Каннингема. Было показано, что у мышей мутации в ДНК-последовательности перестроенного V(D)J-reHa вариабельной области появляются после стимуляции антигеном. Дальнейшие доказательства были получены в новых экспериментах с трансгенными мышами.
Все эти работы стали возможными благодаря разработке в 1975 г. методов «гибридомы» и «моноклональных антител». За создание этих методов Георг Келер и Цезар Мильштейн получили в 1983 г. Нобелевскую премию (совместно с Нильсом Ерне). В этих методах используется слияние раковой клетки (способной расти и неограниченно делиться в культуре) с индивидуальной В-клеткой иммунизированного животного. Гибридная клетка продуцирует антитело В-клетки и приобретает бессмертие раковой клетки. Такие гибридомы позволили экспериментально показать существование клонов В-клеток, предсказанное клонально-селекционной теорией Бернета, причем все потомки одной клетки имеют одинаковые V(D)J-гeны и, таким образом, производят одинаковые антитела. Клон гибридомных клеток дает возможность сравнить ДНК-последовательности соматически перестроенного V(D)J-гена В-клеток и последовательности V-элемента зародышевой линии. Во многих случаях было показано, что В-клетки, изолированные после стимуляции антигеном, накапливают много соматических мутаций в том участке V-элемента, который кодирует последовательность аминокислот от 1-й до примерно 85-й позиции и включает первые два гипервариабельных участка (см. рис. 5.3). Эти данные соответствуют предположению об отборе антигеном в центрах размножения. Частота соматических мутаций очень высока: ДНК-последовательность, включающая перестроенный V(D)J-ген, накапливала изменения, которые отличались от немутантной последовательности зародышевой линии примерно по 5 процентам оснований.
Нам известно, что соматические мутации локализованы в короткой ограниченной области ДНК В-клетки. Экспериментальные данные суммированы на рис. 5.5, где показаны перестроенный VDJ-ген тяжелой цепи в начале реакции в центре размножения и та же последовательность через семь и четырнадцать дней. Обратите внимание, что доля мутаций, по-видимому, увеличивается со временем, и что мутации ограничены ближайшим окружением перестроенного VDJ-гена и некодирующими фланкирующими (расположенными по краям) ДНК-последовательностями. Их не находят около промоторного сай-та, с которым связывается РНК-полимеразный ферментный комплекс и с которого начинается (инициируется) копирование мРНК по матрице ДНК (в кэп-сайте, обозначенном на рисунке маленькой витой стрелкой выше лидерного L-кодирующего участка). Не обнаруживаются они и за участком, который, по нашим предположениям, является «локус-специфичным устройством» (Ei/MAR, intronic Enhancer/Matrix Attachment Region).
В самом VDJ-кодирующем участке мутации, обнаруженные в коллекции последовательностей антител высокой аффинности, распределены не случайно, они демонстрируют «структуру Ву-Кэбота». Таким образом, точковые мутации, которые приводят к замене аминокислоты, накапливаются в гипервариабельных участках (или тех областях белка, которые формируют антигенсвязывающий центр). «Молчащие» точковые мутации (те изменения внутри кодона, которые не приводят к замене аминокислоты, см. приложение) накапливаются в консервативных каркасных участках. Наблюдаемые структуры Ву—Кэ-бота (рис. 5.3) означают, что мутантные антитела подвергаются антигенсвязывающему отбору.
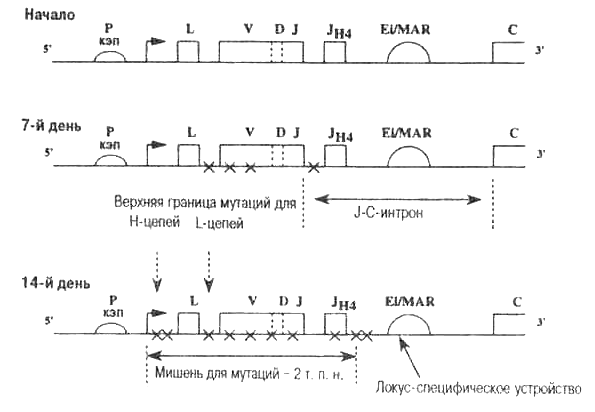
Рис. 5.5. Схема, показывающая, что перестроенный V(D)J-reH и соседние фланкирующие участки ДНК служат мишенью для мутатора.
Изображена тяжелая цепь человека или мыши. Картина мутаций для главного семейства легких цепей (капла-цепей) у человека и мыши одинакова. На рисунке показано, что среди У(0)и-последовательностей, выделенных в позднем иммунном ответе (15 дней после воздействия антигена), выявляется большая доля соматических мутаций. Обратите внимание, что соматические точковые мутации расположены в участке, начинающемся недалеко от точки начала синтеза мРНК (транскрипции) и заканчивающемся в районе J-C-интрона около Ei/MAR. Также обратите внимание, что районы промотора (Р) выше VDJ и ниже константной (С) области не мутируют. Предполагается, что Ei/MAR является локус-спе-цифическим устройством, с которым стыкуется мутаторная машина; Х — точковая мутация, кэп — начало синтеза мРНК; т. п. н.— тысяч пар нуклеотидов. Участки, кодирующие белок, показаны прямоугольниками. Дополнительная информация в табл. 5.1 и на рис. 2.6, 4.5 и 5.1.
Такое распределение иллюстрирует две ключевые черты соматического гипермутирования. Разделение ДНК-последовательностей, кодирующих вариабельные (V) и константные (С) области, дает то эволюционное преимущество, что разрешаются мутации в V-генах, но сохраняются неизменными С-гены. То, что механизм соматического мутирования ограничен только перестроенным V(D)J-геном и не затрагивает гена С-области, особенно важно для тяжелых цепей. Они образуют ту часть молекулы антитела, которая определяет ее функциональные свойства, такие как лизис бактериальных клеток, поглощение и разрушение инфекционных агентов фагоцитами, сигналы В-клеткам к делению и продукции антител, процессы, приводящие к стимуляции Т-клеточной помощи В-клеткам. Для легких цепей этого не требуется, потому что константная область легкой цепи не выполняет этих функций.
Второй принцип, показанный на рис. 5.5, — мутации не распространяются вверх от гена. 5'-граница находится вблизи сайта начала транскрипции или лидерного интрона (некодирующая последовательность между L- и V-кодирующими участками, рис. 4.5). Тому есть очень важная причина, так как в этом участке находятся регуляторные последовательности (промоторный, или Р-район), которые определяют связывание РНК-полимеразного комплекса, инициацию транскрипции и образование мРНК.
Важно отметить, что мутации сосредоточены только там, где они не изменяют ни С-областей, ни Р-районов, которые контролируют экспрессию гена. Как достигается такое точное прицеливание?
Распределение мутаций, показанное на рис. 5.5, и известные уровни ошибок копирования молекул РНК (см. рис. 5.2) были двумя основными фактами, которые привели в 1987 г. Теда Стила и Джеффа Полларда (Pollard) к созданию «модели обратной транскриптазы» для объяснения механизма соматического гипермутирования (для краткости — КТ-модель). Эта идея родилась на год раньше в феврале в Волонгонге и была сформулирована летом 1986 г., когда Тед и Джефф встретились в Нью-Йорк Сити. (К этому моменту оба считали, что поняли почти все, и решили опубликовать свою идею.) Тед считал, что RT-модель следует из теории соматического отбора. Однако аргументы Боба Бландэна убедили Теда, что RT-модель соматического гипермутирования по смыслу должна предшествовать теории соматического отбора. (Также она должна предшествовать и в эволюционном смысле — соматические мутации небольшого начального набора V-генов зародышевой линии должны происходить до передачи информации V-генов от сомы к зародышевой линии. Этот гносеологический поворот на самом деле упрощает интерпретацию данных о ДНК-последовательностях, особенно касающихся генетической рекомбинации V-генов зародышевой линии (см. обсуждение следов интеграции сомы в зародышевую линию и рис. 6.3)
С 1986 г. работа над гипотезой обратной транскрипции продолжалась в нашей лаборатории с участием Джерри Бота (Both) и Гарри Ротенфлу (Rothenfluh). Сейчас мы можем привести детальную теоретическую молекулярную модель соматического гипермутирования в В-клетках: она включает неточную, склонную к ошибкам обратную транскрипцию и возврат генов в ДНК зародышевой линии (рис. 5.6). Эта модель согласуется с подавляющим большинством экспериментальных результатов, касающихся соматического гипермутирования. Ее можно распространить на молекулярные механизмы, которые приводят к соматическому разнообразию перестроенных V(D)J-генов вариабельных областей у кур, до сих пор называемые генной конверсией. Однако мы должны подчеркнуть, что до тех пор, пока все молекулярные детали не будут экспериментально обоснованы, наша модель останется гипотезой, хотя и совместимой со всеми доступными данными.
Мы предположили, что молекулярной машиной, которая с высокой частотой вызывает мутации перестроенной ДНК V(D)J-гена, должна быть «RT-мутаторсома» (RT — обратная транскриптаза). Существует много молекулярных органелл с суффиксом «сома», например «рибосома» (комплекс белков и РНК, необходимый для трансляции информационной РНК в последовательность аминокислот, см. приложение) и «сплай-сосома» (также РНК-белковый комплекс, который вырезает интроны из про-мРНК). Итак, гипотетическая RT-мутатор-сома использует несплайсированную про-мРНК как матрицу для синтеза кДНК. Термин кДНК, где «к» обозначает комплементарная — общий термин для всех ДНК-копий РНК-матрицы, созданных обратной транскриптазой (кДНК также называют «обратными транскриптами» или «ретротранскриптами».
Мы предположили, что обратная транскрипция, которая создает мутантную кДНК-копию перестроенного V(D)J-yчастка, начинается в особом районе, в «праймерном» сайте ниже V(D)J около Ei/MAR участка (рис. 5.6), и продолжается справа налево по направлению к кэп-сайту (5'-конец про-мРНК матрицы). Цезар Мильштейн с коллегами экспериментально показали на трансгенных мышах, что «локус-специфическое устройство», Ei/MAR, важно для соматического гипермутирования, тогда как V(D)J-кодирующий участок и промотор можно заменить копиями гемоглобинового гена без ущерба для мутации. Мы считаем Ei/MAR «локус-специфичным устройством», необходимым для стыковки RT-мутаторсомы с V(D)J-геном и ограничения мутаций этим геном. (В настоящее время мы экспериментально проверяем это предположение.) Мы также считаем, что мутантная кДНК-копия V(D)J-yчастка встраивается в хромосому и замещает исходный, немутированный V(D)J (на рисунке это показано петлеобразной стрелкой.). Возможность подобной генетической интеграции экспериментально продемонстрирована у многих организмов и называется гомологичной рекомбинацией, так как похожие ДНК-последовательности совмещаются, а за этим следует рекомбинация ДНК. Указанные предположения гарантируют, что участки выше промотора и ниже константного участка защищены от мутаций, а некодирующая ДНК в непосредственном соседстве с V(D)J мутирует с очень высокой частотой (тот же уровень ошибок, что и при транскрипции и обратной транскрипции — примерно 10-3—10-4 на цикл копирования пар оснований, рис. 5.2)
Таким образом, правила копирования ДНК- или РНК-матриц и склонные к ошибкам процессы синтеза РНК и кДНК полностью удовлетворяют «требованиям» соматического мути-рования. Существует единственное направление, в котором могут синтезироваться ДНК-копии по матрице про-мРНК — обратно к сайту начала транскрипции (кэп-сайту). Если синтез кДНК начинается в Ei/MAR-участке или рядом с ним, это автоматически обеспечит мутирование V(D)J без риска мутирова-ния промотора и константного участка. Для того чтобы «обессмертить» мутантную последовательность в организме, потребуется гомологичная рекомбинация для встраивания мутант-ной кДНК-копии в хромосомную ДНК, что обеспечит передачу ее последующим поколениям дочерних клеток.
У мышей 5', или верхняя, граница мутаций находится около кэп-сайта для Н цепей и в L-V интроне для легких цепей (рис. 5.5). Расположение этих сайтов согласуется с двумя главными точками, где заканчивается синтез кДНК, а) когда обратная транскриптаза подходит к 5'-концу матрицы про-мРНК, или б) около L-V-интрона, так как интрон может быть удален при сплайсинге, который превращает про-мРНК в мРНК.
Какие еще данные свидетельствуют в пользу RT-модели и отличают ее от других мутационных моделей, которые мы определяем как «основанные на ДНК» (они зависят от локального склонного к ошибкам синтеза ДНК вблизи перестроенного V(D)J-участка)? Прежде всего, одно общее соображение: нет доказательств существования механизма, который избирательно прекращает синтез ДНК. Однажды начавшись, синтез продолжается до тех пор, пока не достигнет конца матрицы. Таким образом, для подтверждения таких моделей потребовалось бы придумать и экспериментально доказать особые правила синтеза ДНК.
Есть еще две группы данных, которые не соответствуют моделям, основанным на ДНК, но которые предсказываются RT-моделью. Они получены в лабораториях Патрисии Гир-харт и Эрика Сейсинга (Seising). В одних экспериментах сразу ниже перестроенного V(D)J-участка (между VDJ и J на рис. 5.6) была помещена так называемая «репортерная» последовательность. Оказалось, что эта последовательность подавляла появление мутаций в VDJ-участке. Такой результат несовместим с моделями, основанными на ДНК, но согласуется с RT-моделью. Действительно, репортерная последовательность — короткий участок ДНК, кодирующий транспортную РНК (тРНК), которая складывается в характерную трехмерную форму (транспортные РНК участвуют в синтезе белка, они переносят аминокислоты). Согласно RT-модели, синтез кДНК должен остановиться до V(D)J-участка, поскольку обратная транскриптаза не может продолжить движение через тРНК-структуру. Два других свойства тРНК также могут прекратить обратную транскрипцию. Первое, последовательность тРНК могла высвободиться из про-мРНК и, таким образом, РНК-матрица, по которой RT-мутаторсома копирует РНК в кДНК, обрывается. Второе, химическая модификация азотистых оснований в тРНК может подавлять синтез кДНК RT-мутаторсомой.

Рис. 5.6. RT-мутаторсома. Изображена тяжелая цепь человека или мыши. Предполагается, что RT-мутаторсома работает одинаково у человека и мыши в случае главного семейства легких цепей (таппа-цепей). События копирования и рекомбинации, изображенные на рисунке, происходят в ядре мутирующей В-клетки. Локус-специфическое устройство (Ei/MAR) стыкуется с RT-мутаторсомой. Поэтому обратная транскрипция матрицы про-мРНК начинается выше Ei/MAR, но ниже V(D)J. Это достигается благодаря тому, что все синтезы нуклеиновых кислот (или полинуклеотидов) всегда идут в направлении 5'— 3'. Значит, все копируемые матрицы должны иметь антипараллельную ориентацию 3'—5'. Матричная цепь ДНК для синтеза РНК — это 3'—5'-цепь. В кэп-сайте (cap site) начинается синтез про-мРНК, и, поскольку это склонный к ошибкам процесс, очень велика вероятность того, что транскрибированная копия V(D)J-участка будет нести замены оснований (обозначено X). Обратная транскрипция, которая также склонна к ошибкам, начинается на про-мРНК выше Ei/MAR и продолжается по направлению к 5'-концу про-мРНК. Мутантный ретротранскрипт (кДНК) гомологично встраивается в ДНК (показано дугообразной стрелкой) и замещает исходную немутантную У(0)и-последовательность. Затем мутантные РНК-транскрипты, подвергаются процессингу (вырезаются интроны) и экспортируются в цитоплазму, где они транслируются в Н- и L-цепи, из которых образуется белковое антитело (Ig), которое проверяется на поверхности В-клетки на связывание с антигеном, презентируемым ФДК. Высокая аффинность к антигену подает стоп-сигнал мутированию. Дополнительную информацию об обозначениях и идеях можно найти в табл. 5.1 и рис. 4.5, 5.4 и 5.5. (По Steele E. J., Rothenfluh H. S., Blanden R. V. Immunology and Cell Biology, vol. 75: 82-95, 1997.)
Другие эксперименты касаются гомологичной рекомбинации. В опытах на трансгенных мышах, имеющих два немного различающихся тесно сцепленных перестроенных V(D)J-reHa, было показано, что соматическая точковая мутация в V(D)J всегда приводит к гомологичной рекомбинации с другими тесно сцепленными У(0).?-последовательностями. Обязательная связь соматической точковой мутации с гомологичной рекомбинацией — поразительное открытие. Этот результат не имеет смысла в рамках мутационных моделей, основанных на ДНК, так как в них гомологичная рекомбинация не играет никакой роли. Однако в RT-модели гомологичная рекомбинация — неотъемлемая составная часть мутационного процесса (рис. 5.6). Таким образом, в хромосомной ДНК-последовательности точковая мутация не возникнет до тех пор, пока мутантная кДНК не рекомбинирует, заменив исходную немутантную V(D)J-пoследовательность.
Следовательно, в настоящее время теория мутаций, основанная на обратной транскрипции, дает лучшее объяснение всем существующим данным о соматическом гипермутиро-вании. Предложенный более 10 лет назад, этот механизм не опровергнут ни одним из многочисленных экспериментов, проведенных с тех пор. Он прошел, как сказал бы покойный философ науки сэр Карл Поппер (Popper), «суровые испытания».
В начале этой главы, мы задались вопросом: «Каков механизм предотвращения лишних, портящих успешно отобранную последовательность, мутаций?» Ответ должен быть таким: селективное преимущество (в традиционном дарвиновском смысле) состоит в том, что когда антитело в центре размножения изменится настолько, что будет обладать высокой аффинностью к антигену, В-клетке должен быть дан сигнал остановить дальнейшее мутирование. Тогда мутаций не будет слишком много, и риск потери высокой аффинности к антигену будет минимизирован. Мы, следовательно, предполагаем, что сигнальная функция поверхностного Ig-рецептора была отобрана в ходе эволюции. Если мутантная В-клетка успешно связывает антиген, присутствующий на фолликулярной дендритной клетке (ФДК) в форме комплекса антиген—антитело, передается СТОП-сигнал для выключения процесса соматического мутирования (рис. 5.6). Этим сигналом может быть прекращение производства белковых субъединиц RT-мутаторсомы (белки постоянно разрушаются ферментами, называемыми протеаза-ми. Следовательно, они исчезают до тех пор, пока не начнется новый синтез).
Позволим себе коснуться другого важного момента, который молчаливо властвует в размышлениях о соматических мутациях. Все мутационные модели, основанные на ДНК, зависят от клеточных делений. Необходимым условием возникновения мутаций считается репликация ДНК и, следовательно, деление клеток. (Считается, что мутации возникают в результате ошибок репарации ДНК.) Сейчас предполагают, что мутации генерируются в фазе быстрого деления клеток (в центробластах), которая дает большую популяцию В-клеток (центроцитов) в центре размножения (рис. 5.4). Однако RT-модель не зависит ни от репликации ДНК, ни от клеточного деления — мутации могут возникать в отсутствие этих двух процессов. Репликация ДНК требуется только для размножения мутантной V(D)J-пoследовательности в делящихся В-клетках. Деление В-клеток, производящих антитела высокой аффинности, полезно, но деление мутантных В-клеток, которые потеряли аффинность к антигену, слишком расточительно.
Представление, что «клеточное деление необходимо для мутирования», настолько глубоко укоренилось в сознании многих, что скорость соматического мутирования записывают как «10-3 - 10-4 на основание на поколение». Другими словами, в традиционное описание скорости мутирования встроена идея клеточного деления, и она невольно оказывает влияние на представления о механизмах мутирования.
Так как RT-модель не зависит от клеточных делений, мы предлагаем циклический процесс «мутация — пауза (для экспрессии Ig) — проверка аффинности», протекающий в неделящейся транскрипционно активной клетке, которая продуцирует мРНК и белковые молекулы Н- и L-цепей антитела и компоненты RT-мутаторсомы. Этот путь организации процесса мутаций и отбора максимизирует образование антител высокой аффинности. Он более эффективен, чем модели, основанные на клеточных делениях.
Мы описали протекающий в В-лимфоцитах процесс обратной связи V(D)J-генов, основанный на склонной к ошибкам обратной транскрипции. Он составляет основу определяемого антигеном мутирования генов антител. Все экспериментальные данные согласуются с этой теорией. Это приводит нас ко второй неоламаркистской концепции: «направленной» обратной связи генов.
Быстрое случайное мутирование, приводящее к различным измененным ДНК-последовательностям, и отбор среди них лучшей последовательности приводят к появлению «направленных соматических мутаций» — полной противоположности случайных мутаций. Играет ли эта соматическая генетическая изменчивость какую-нибудь роль в эволюции? Точнее, можно спросить: есть ли какие-нибудь свидетельства того, что приобретенные соматические мутации генов вариабельных областей могут вносить вклад в следующее поколение? Другими словами, могут ли приобретенные соматические мутации наследоваться с ДНК половых клеток? Может ли предполагаемая нами гомологичная рекомбинация приводить к переносу V-последовательности из В-лимфоцита в ДНК сперматозоидов или яйцеклеток?